-
Home


-
Our research
-
Current research
-
Our IRD funded research
- Dr Forbes Manson, University of Manchester
In search of new drug therapies for inherited retinal diseases
- Grant holder: Dr Forbes Manson, Senior Lecturer in Ophthalmology with PhD student Kathleen Elverson
- Institution: University of Manchester
- Grant award: £65,021
- Start date: January 2019
- End date: April 2022
Why is this research needed?
Inherited retinal diseases (IRDs) are the commonest cause of blindness amongst working age adults in the UK, and the second most common cause in children. They are caused by mutations in our genetic code and there is currently no cure or specific treatment available.
Genetic mistakes that result in misfolded protein products may be responsible for up to half of all human diseases – including inherited retinal diseases. Proteins are chains of building blocks (amino acids) that have to fold correctly in order to function normally. Genetic mistakes that cause the wrong amino acid to be inserted prevent proper protein folding, resulting in proteins being non-functional or recycled. Chemicals called chaperones can help abnormal proteins to fold properly and regain their function.
Dr Manson’s group works on inherited retinal diseases caused by mistakes in the BEST1 gene that makes a protein called bestrophin-1, a protein channel in the retinal pigment epithelium (RPE) of the retina. The RPE is a single layer of highly pigmented cells that performs many critical functions, including supporting retinal photoreceptors, which convert light into signals that are sent to the brain, enabling us to see.
Over 120 different mistakes in BEST1 cause eye disease, and most cause bestrophin-1 to mis-fold, resulting in a loss of the protein’s function. Normally, bestrophin-1 controls the exit of chemicals (chloride ions) from a cell, like a gate that only allows a particular chemical through. One of the diseases caused by mistakes in bestrophin-1 is called autosomal recessive bestrophinopathy (ARB). Faulty ARB bestrophin-1 does not allow chloride ions to leave the cell, similar to the gate being the wrong shape and jammed shut. Dr Manson’s research team has previously used a chaperone called 4PBA to help faulty ARB bestrophin-1 proteins regain their shape and function. However, researchers had to use more 4PBA than is ideal for a therapeutic drug.
What is the aim of the project?
The ultimate aim of this project is to identify drug candidates that are more suitable than 4PBA.
The project has two main parts. Firstly computer modelling is being used to identify the location of mutations in bestrophin-1 and also to investigate the binding of chemicals to a protein called COPII that regulates how a cell processes mutant forms of bestrophin-1. The second part of the project uses experiments to test whether chemicals identified by computer modelling have a beneficial effect on the function of mutant bestrophin-1.
As a first step, Dr Manson’s team has developed a model of the target human protein (bestrophin-1) using a chicken protein template. They modelled where the mutations in bestrophin-1 occurred to cause two diseases: BVMD (Best vitelliform macular dystrophy) and ARB (autosomal recessive bestrophinopathy). They found that the mutations that cause each disease are grouped and differently distributed from one another. This may explain why different mutations cause different diseases.
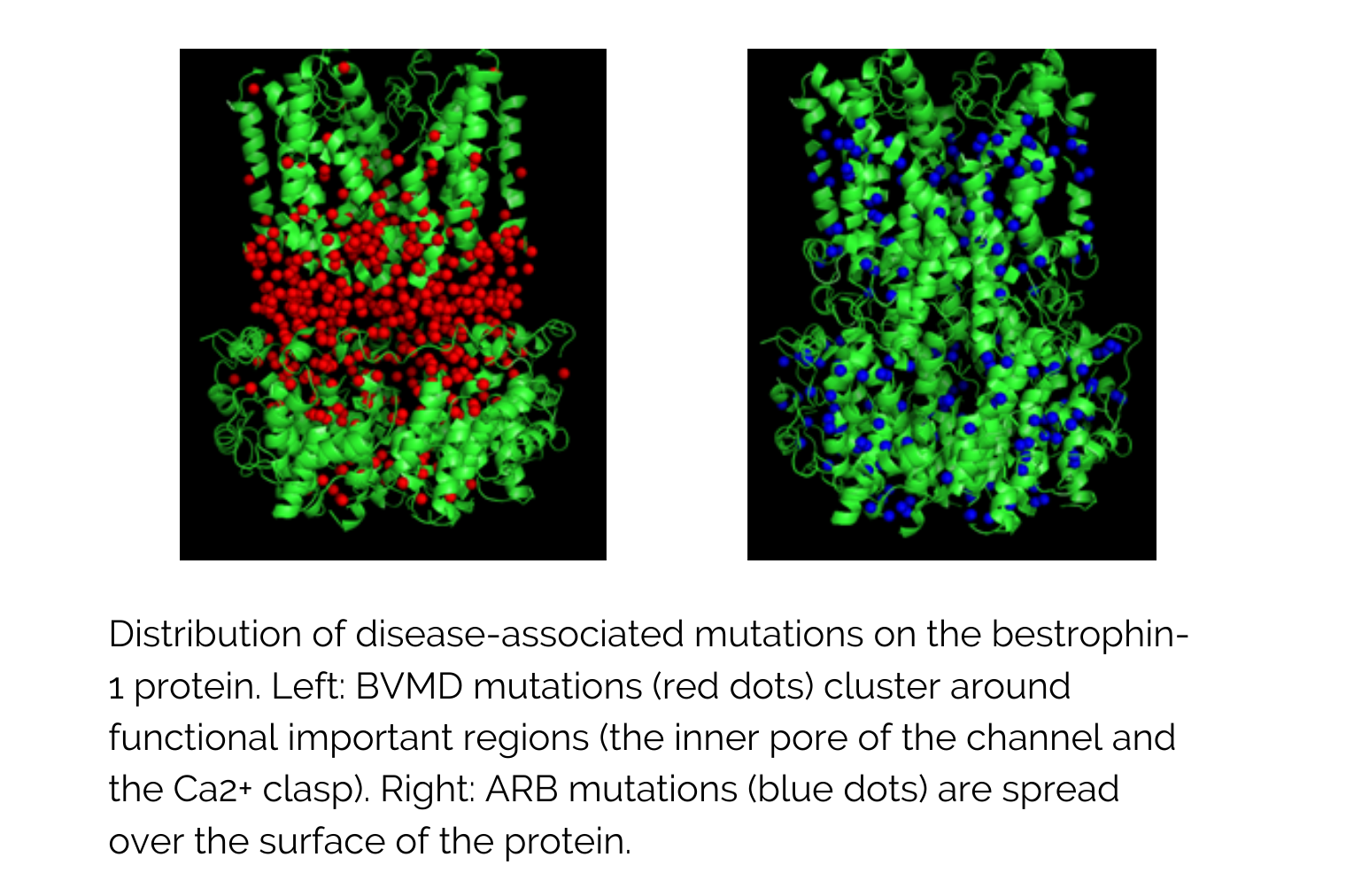
How will this research help to beat sight loss faster?
Restoring the function to misfolded proteins that cause inherited retinal diseases has the potential to prevent the onset of diseases, reduce their severity, or halt their progression. This approach could also be used for other eye diseases that are caused by misfolded proteins.
Thank you
In 2017, Sight Research UK launched a special appeal in support of Dr Manson’s project. His research is being made possible thanks to the generosity of our community of donors. In particular, we would like to thank the Hobson Charity, the James Tudor Foundation, the Charles Wolfson Charitable Trust, the P F Charitable Trust, the Sir Samuel Scott of Yews Trust, the Dorothy Hay-Bolton Charitable Trust, the Payne-Gallwey Charitable Trust, the Hospital Saturday Fund, the Douglas Heath Eves Charitable Trust, and the Roger Raymond Charitable Trust for their significant contributions.
Further information
The most common type of IRD is retinitis pigmentosa. You can find more about the causes and symptoms of this condition here.
Stop the clock on sight loss
Every 6 minutes someone in the UK receives the devastating news that they are going blind. That’s 250 people a day.
Your gift can help to find new sight-saving solutions.
If you can, please donate today. Thank you.
Read next
Professor Gavin Arno, UCL